
A lan Aspuru-Guzik from Harvard gave a very nice lunchtime seminar at MIT recently on using quantum computers for the simulation of chemical systems. The key point Alan made is that chemistry provides a wealth of problems that seem to provide the most immediate practical prospects for the demonstration of quantum speed-ups. His assessment is that the simulation and energy estimation of a chemical molecule (the energy being the key quantity required to estimate reaction times) made of about 100 atoms is well beyond the reach of classical simulation techniques (even approximate and heuristic), but that such a simulation could be performed on a quantum computer with roughly the same number of qubits. A major advantage seems to be that there is not much overhead in the number of qubits required to simulate the molecule, compared to the size of the molecule itself (as opposed to, say, factoring a 100-digit number).
lan Aspuru-Guzik from Harvard gave a very nice lunchtime seminar at MIT recently on using quantum computers for the simulation of chemical systems. The key point Alan made is that chemistry provides a wealth of problems that seem to provide the most immediate practical prospects for the demonstration of quantum speed-ups. His assessment is that the simulation and energy estimation of a chemical molecule (the energy being the key quantity required to estimate reaction times) made of about 100 atoms is well beyond the reach of classical simulation techniques (even approximate and heuristic), but that such a simulation could be performed on a quantum computer with roughly the same number of qubits. A major advantage seems to be that there is not much overhead in the number of qubits required to simulate the molecule, compared to the size of the molecule itself (as opposed to, say, factoring a 100-digit number).
The idea of using quantum comput ers for the simulation of quantum systems famously goes back to Feynman, who is usually credited as the first to envision the possibility for a universal quantum computer. Before Alan’s talk I never paid too much attention to this statement, which sounded like a straightforward extension of the universality of the Turing machine. But if you take a moment to think about what it really means, it is quite a bold statement: there exists a certain fixed physical system which is capable of simulating the dynamics of any other physical system. That this would be such a feat struck me when Alan mentioned that the first simulation of a chemical system was obtained by simulating a hydrogen molecule
ers for the simulation of quantum systems famously goes back to Feynman, who is usually credited as the first to envision the possibility for a universal quantum computer. Before Alan’s talk I never paid too much attention to this statement, which sounded like a straightforward extension of the universality of the Turing machine. But if you take a moment to think about what it really means, it is quite a bold statement: there exists a certain fixed physical system which is capable of simulating the dynamics of any other physical system. That this would be such a feat struck me when Alan mentioned that the first simulation of a chemical system was obtained by simulating a hydrogen molecule 
A photon (sort of)
An H2 molecule (sort of)
quantum dynamics of an electron orbiting a proton! This may sound naïve, but the fact that nature would allow for such universality strikes me as entirely non-obvious. I recommend a very nice article by Seth Lloyd for a thorough discussion of Feynman’s suggestion; reading it gives a good idea of the many ways in which this could have failed (and still could).
Alan made a number of interesting points in his talk. The first is that, in terms of complexity the theoretical bottleneck of a simulation procedure, in terms of complexity, seems to be its first step, the state preparation (the other steps being the evolution and the measurement). Given a certain molecule and a target system in which to encode its wavefunction, how does one actually prepare the target in the appropriate state? Typically we will be trying to encode the ground state of the molecule under some Hamiltonian. Solving for a complete description of that state could be a hard problem — there could be exponentially many parameters to determine. As pointed out by the audience, the standard heuristic argument that this should be a non-issue can be made: if the molecule exists in nature, certainly it can be prepared efficiently (since nature did it). In practice, this heuristic suggests that adiabatic evolution would be a good method to perform the preparation. But the efficient preparability (I know…) of the original molecule does not seem sufficient: what is really needed is to initialize the target system used to represent the molecule. The dynamics of that system may be quite different, and implementing the Hamiltonian driving it towards the state chosen to represent the molecule’s ground state may not be as easy. Here it seems crucial to choose an appropriate representation, and not any representation: again, determining such a representation may not be an easy problem in general.
I mentioned above that the simulation of a 100-atom molecule could roughly be done using a 100-qubit computer. There is a catch: implementing the molecular evolution operator, which will drive the initial state towards the state whose energy we wish to estimate, may require a huge numbers of gates (Alan mentioned the number 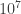
 what is currently achievable — but is well below the number of gates that your CPU applies per second). There is a trade-off: the most efficient encoding might not lead to the simplest simulation. That the evolution operator would be implementable using only a polynomial number of quantum gates is itself non-trivial (it is, after all, an exponential-sized matrix) but owes to the fact that it is typically local: the sum of terms acting only on spatially neighboring atoms (see Lloyd’s survey referred above for more on this).
what is currently achievable — but is well below the number of gates that your CPU applies per second). There is a trade-off: the most efficient encoding might not lead to the simplest simulation. That the evolution operator would be implementable using only a polynomial number of quantum gates is itself non-trivial (it is, after all, an exponential-sized matrix) but owes to the fact that it is typically local: the sum of terms acting only on spatially neighboring atoms (see Lloyd’s survey referred above for more on this).
In spite of these difficulties Alan is quite optimistic at the prospects for chemical simulations, and he invited us computer scientists to demonstrate the computational hardness of the problems he is trying to solve. The goal is to obtain a good theoretical basis for the demonstration of a quantum speed-up, were these simulations to be carried out. At this point the audience challenged him a little bit: indeed, as compared to, say, computational biology, there are currently very few researchers with a training in computer science working on the kinds of problems Alan mentioned. Are they really hard? Years of concerted attempts at finding efficient simulations and heuristic algorithms seems to suggest so; on the other hand computer science does have more than one trick up its sleeve, and some of the most powerful algorithmic methods developed in recent years (spectral methods, semidefinite programs, etc.) may not be well-known to chemists.
I was never much into chemistry, but this talk managed to drive up my enthusiasm by a few orders of magnitude (I won’t stay where I started the day :-)). I wouldn’t have thought that the prospect for simulating a molecule would sound so much more exciting than that of factoring a 100-digit number, but right now it does! If anyone is interested in reading more about the complexity-theoretic aspects of Alan’s work I highly recommended the recent survey by James Whitfield, Peter Love and Alan, which even though written for an audience of chemists gives a great account of the kinds of computational problems that arise in performing chemical simulations.

Nice blog! Is your theme custom made or did you download it from somewhere?
A design like yours with a few simple tweeks would really make my blog stand out.
Please let me know where you got your theme. Cheers
Thanks. I think the theme is a standard WordPress theme.
Aspuru Guzik’s amazing scientific originality never ceases to astound me. That review that you just mentioned. When I read it, it crossed my mind that these guys had learned their complexity theory by reading solely Wikipedia. But then I thought further and realized that this was classic Aspuru, trying to dress down his vast knowledge of complexity theory so that his review was accessible to the layman. Just today, Aspuru astounded me again with his amazing originality by presenting a one line (but very in depth) analysis of your blog post. The richness of Aspuru’s blogging puts yours to shame. But that is to be expected since he hails from Harvard and you only from MIT.
Pingback: IBM Introduces the World to Quantum Cloud Computing – Ahead in the Cloud